
The use of light as an external stimulus to achieve reversible gas capture was demonstrated for the first time in 2012 in a proof of concept work by Park and coworkers using azobenzene-functionalized metal-organic frameworks [1]. This work represented an important contribution in the field because the use of light rather than temperature potentially enables a more energy-efficient separation process. However, although this strategy possesses great promises towards separation applications, the development of the materials remains slow due to the challenge in synthesizing functionalized materials that can accommodate reversible isomerization of the photoactive moieties. Also, limited atomistic studies on the adsorption mechanism in these materials are available.
In the context of the ANR JCJC ComputationalCarbonCapture [2015-2020], we have developed a strategy allowing us to couple different atomistic computational methods in order to understand the thermodynamics of carbon capture by photo-active metal-organic frameworks and their excited state properties. Specifically, as part of a long-lasting collaboration with Prof. Lin at Ohio State University, we have coupled first principle calculations and classical molecular simulations to explain, for the first time, the reversible gas uptake by two families of photo-active MOFs [1,2].
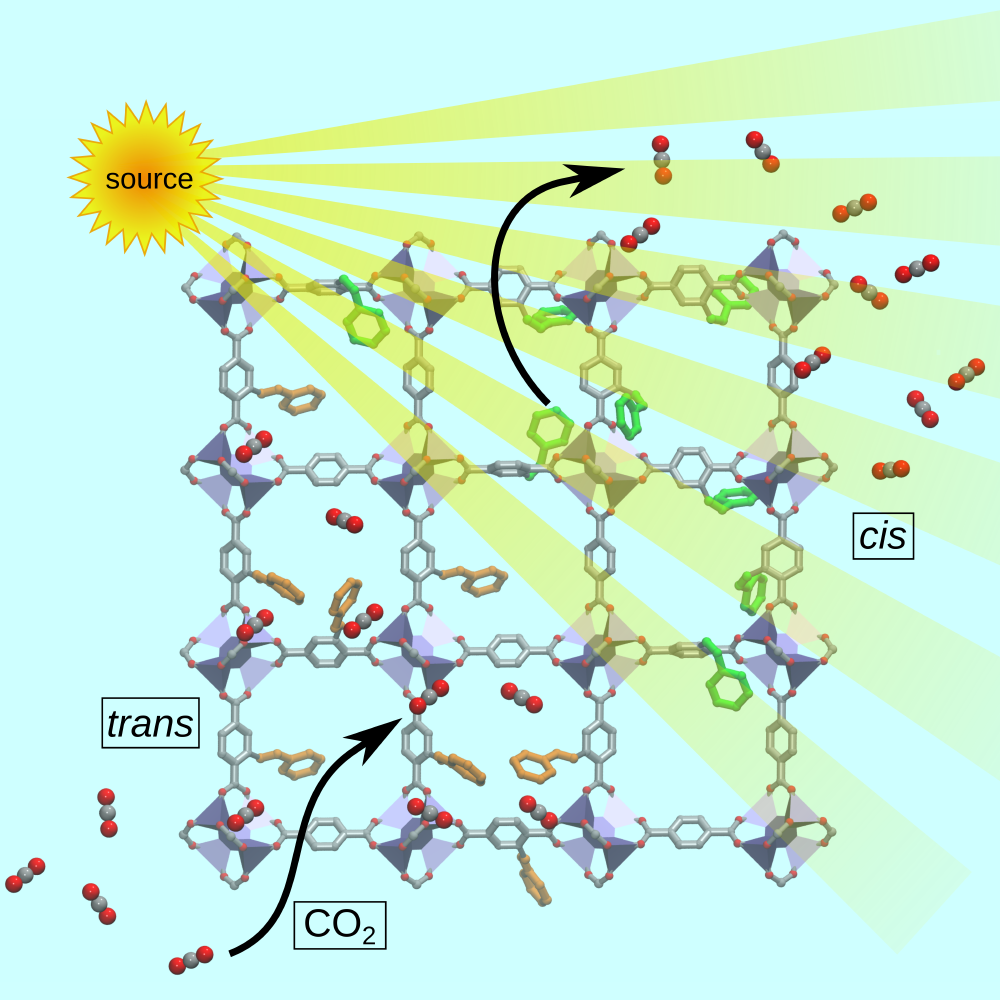
Tuning Gas Adsorption by Metal Node‐Blocking in Photoresponsive Metal‐Organic Frameworks C-T. Yang, A. R. Kshirsagar, A. Charaf-Eddin, L.-C. Lin, R. Poloni, Chemistry: a European Journal 24, 15167 (2018), Editor’s choice: Hot Paper;
Tuning Gas Adsorption by Metal Node‐Blocking in Photoresponsive Metal‐Organic Frameworks C-T. Yang, A. R. Kshirsagar, A. Charaf-Eddin, L.-C. Lin, R. Poloni, Chemistry: a European Journal 24, 15167 (2018), Editor’s choice: Hot Paper;
In this study we demonstrated that the reversible decrease in gas adsorption upon isomerization can be primarily attributed to the blocking of the strong adsorbing sites at the metal nodes by azobenzene molecules in cis configuration, as illustrated in the Figure. We found the same mechanism to apply to other molecules, for example, alkanes and toxic gases and we anticipate a similar behavior in most frameworks exhibiting exposed (an not necessarily open) metal centers.
Because the identified mechanism implies that larger fractions of cis would yield a larger working capacity, we are now studying the optical absorption properties of azobenzene-functionalized MOFs using CASSCF/CASPT2 and the Bethe-Salpether formalism.
Contact: Roberta Poloni
Contact: Roberta Poloni