
Gaining a better understanding of the properties and behaviour of materials requires a precise description of the mechanisms involved at several spatial and temporal scales.
Modelling from atomic scale upwards on the basis of Density Functional Theory enables any type of material to be explored at different spatial and temporal scales ranging from the electronic structure to bulk materials. The approach adopted by the TOP group, known as in silico, is predictive insofar as it begins by taking into account the electronic structure from first principles without relying on experimental information.
Throwing light on the undercooling puzzle
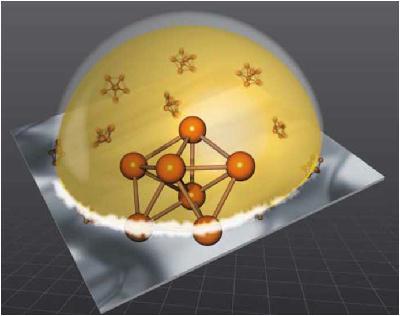 | Using ab initio molecular dynamics simulations, a new remarkable undercooling phenomenon has been explained, namely an undercooling as deep as 350°C for Gold-Silicon eutectic alloy in contact with a specially decorated silicon (111) surface where the outermost layer of the solid featured pentagonal atomic arrangements. Such a five-fold coordinated surface influences the short-range order and the metastability of the liquid, favoring the existence of pentagons in this phase. This result has wide implications not only for fundamental studies of freezing, but also for practical control of the phase transition (more). (coll. ESRF, RTRA Nanosciences, TU Wien, Contacts: A. Pasturel , N. Jakse) |
Atomic scale
The methodology used at atomic scale is based on molecular dynamics (MD), providing access to the structural, dynamic and magnetic properties as well as electronic and atomic transport characteristics. A powerful method involves combining MD directly with the interatomic forces derived from the DFT [Phase Transition, 80, 369 (2007)]. A hybrid technique, associating the speed of conventional MD and the precision of MD ab initio, has proved to be essential for studying the liquid-liquid transition of silicon [Phys. Rev. Lett. 99, 205702 (2007)].
Microscopic scale
At macroscopic level, we are able to study the appearance of base states for binary and ternary systems and their progress depending on temperature, pressure or stress, by combining energy calculations derived from the ab initio approaches with entropy calculations based on statistical physics. In the case of multi-component systems with a high industrial impact, we are developing more versatile techniques combining ab initio calculations with CALPHAD methods.